Overview:
Research Topics of CBDD Group
- Chemoinformatics
- Bioinformatics
- Drug Design
- Chemo- and Geoinformatics
- Webserver and database
- Machine Learning
TargetNet is a open web server that could be used for netting or predicting the binding of multiple targets for any given molecule.
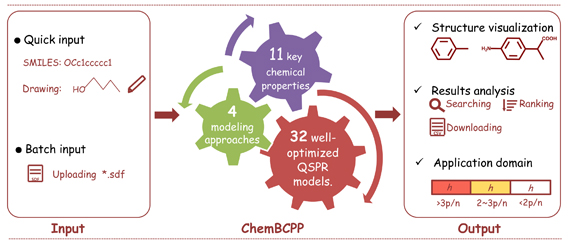
ChemBCPP: An online platform for predicting basic chemical properties
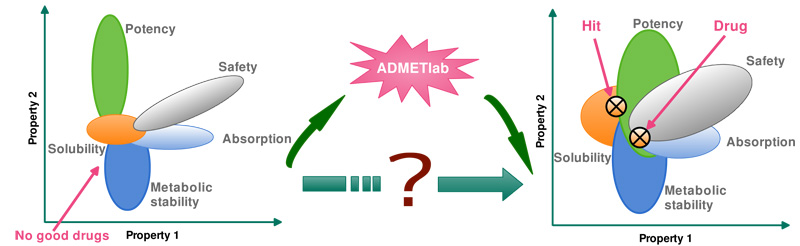
ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database.
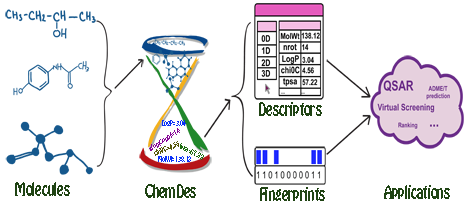
ChemDes:An online-tool for the calculation of molecular descriptors.

ProtrWeb:Calculate Protein Sequence-Derived Structural and Physicochemical Descriptors.
Tools and packages:
We have developed a series of software and web servers that can assist in cheminformatics and drug discovery. The tools are listed as following:
-
BioMedR
Jie Dong, Min-Feng Zhu, Dong-Sheng Cao, et al. Briefings in Bioinformatics 2019
-
ChemDes
Jie Dong, Dong-Sheng Cao, Hong-Yu Miao, et al. Journal of Cheminformatics 2015, 7:60
-
ProtrWeb
Nan Xiao, Dong-Sheng Cao, MinFeng Zhu, et al. Bioinformatics, 2015, 31(11): 1857-1859.
-
BioTriangle
Jie Dong, Zhi-Jiang Yao, Ming Wen, et al. Journal of Cheminformatics 2016, 8:34
-
TargetNet
Zhi-Jiang Yao , Jie Dong , Yu-Jing Che, et al. Journal of Computer-Aided Molecular Design, 2016, 30(5): 413-424.
- ADMETlab
Jie Dong, Ning-Ning Wang, Zhi-Jiang Yao, et al. Journal of Cheminformatics, 2018, 10:29
-
ChemBCPP
Dong J, Wang N N, Liu K Y, et al. Chemometrics and Intelligent Laboratory Systems, 2017, 171:65-73.
-
ChemSAR
Jie Dong, Zhi-Jiang Yao, Min-Feng Zhu, et al. Journal of Cheminformatics. 2017, 9(1): 27.
-
PyBioMed
Dong J, Yao Z J, Zhang L, et al. Journal of cheminformatics, 2018, 10(1): 16.
-
ECoFFeS
Liu Z Z, Huang J W, Wang Y, et al. IEEE Access, 2018, 6: 20950-20963.
-
HAMdb
Ning-Ning Wang, Jie Dong, Lin Zhang, et al. Journal of Cheminformatics, 2018, 10:34
-
Chemopy
Cao D S, Xu Q S, Hu Q N, et al. Bioinformatics, 2013, 29(8): 1092-1094.
-
Rcpi
Cao D S, Xiao N, Xu Q S, et al. Bioinformatics, 2014, 31(2): 279-281..
-
PyDPI
Cao D S, Liang Y Z, Yan J, Journal of Chemical Information and Modeling.2013, 53(11): 3086-3096
-
propy
Cao D S, Xu Q S, Liang Y Z. Bioinformatics, 2013, 29(7): 960-962.
-
AlzheimerNet
submitted
-
rDNAseWeb
submitted
-
GPCRnet
submitted
-
PyNetSim
submitted
Recent Projects:
- metaTarFisher tries to fish the credible target of a compound. Target finding is one of the most important challenges in drug discovery. Some good methods and tools have been established to obtain the possible targets of a compound. However, usually only part of the information can be captured. Here, we develop metaTarFisher that tries to grasp comprehensive information of the target by integrating state-of-art target searching tools. We hope metaTarFisher can be used as a meta target searching tool.
- Deep learning models used to establish new target prediction models.
- More than 10 popular target prediction tools were integrated.
- Rich target prediction reports and clear visualization.
- The current version of BioMedR could calculate 293 small molecular descriptors and 13 kinds of molecular fingerprints, 9920 protein descriptors based on protein sequences and six types of generalized scale-based descriptors for proteochemometric modeling, more than 6000 DNA descriptors from nucleotide sequences, and six types of interaction descriptors using three different combining strategies.
- This package realized 5 similarity calculation methods and 4 powerful clustering algorithms as well as several useful auxiliary tools, which aims at building an integrated analysis pipeline for data acquisition, data checking, descriptor calculation and data modeling.
- TCMSID, a Traditional Chinese Medicine Simplified Integrated Database, which contains 499 herbs registered in the Chinese pharmacopoeia with 17920 ingredients including comprehensive annotations, was developed to achieve TCM Simplification. In this database, several key ingredients can be screened as representatives of the whole TCM to capture potential targets via implementing multi-tool target prediction. Therefore, Networks among TCM herbs, formulations, ingredients, potential targets can be constructed to facilitate clarifying function and mechanisms of the TCM.
- Constructed a Traditional Chinese Medicine Simplified Integrated Database containing 17246 herbal ingredients;
- Evaluated 14 kinds of ADME/T properties for all ingredients;
- Provided potential targets of ingredients by multi target prediction tools;
 湘ICP备15000132号-1
湘ICP备15000132号-1 湘公网安备 43010402000120号
湘公网安备 43010402000120号